Haplogroup estimation
The mtDNAmanager provides a web-based forensic mitochondrial bioinformatics resource for supporting data quality control based on haplogroup estimation. The prediction of the most probable haplogroup of a given mtDNA control region sequence data is implemented using a mathematical algorithm based on propositional logic via a hierarchical checking of the presence or absence of haplogroup-specific diagnostic mutations. For that purpose, we first identified the reliable
control region mutation motifs for the assignment of more than
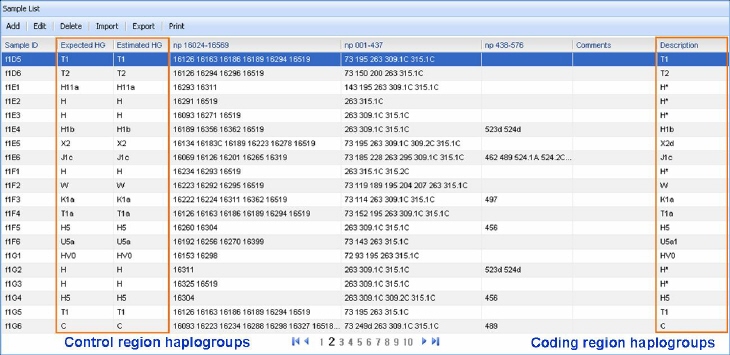
630 mtDNA haplogroups and subhaplogroups based on the well characterized global mtDNA phylogeny. Then, we weighed the individual diagnostic positions on each haplogroup backgrounds supported by datasets containing control region sequences, whose haplogroup-affiliation was confirmed by coding region SNPs. Generally, very good concordance is observed between control region and coding region haplogroups. Actually, using the bioinformatics tools of mtDNAmanager, more than 98% of mtDNAs of high quality datasets can be allocated to the same mtDNA haplogroups as those confirmed with the coding region SNP information .
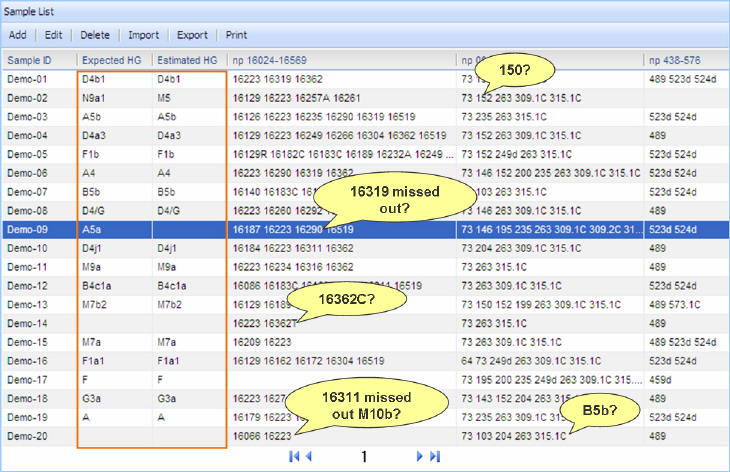
Especially, as mtDNAmanager took account of the different mutation rates of the individual diagnostic positions on the different haplogroup backgrounds, it facilitates mtDNA data quality control. That is, mtDNAmanager allows the respective check of the presence of key diagnostic mutation motifs and accompanying mutations by designating both "expected" and "estimated" haplogroups for each mtDNA sequence. Accordingly, if a certain mtDNA sequence is annotated only with the expected haplogroup, it suggests the lack of accompanying mutations for the annotated expected haplogroup which is determined by the presence of the key diagnostic mutation motifs. From a mtDNA phylogeny point of view, it means that a given mtDNA haplotype is located at a previously unsampled interior node of the tree. Accordingly, there is a need to recheck the presence or absence of the whole haplogroup-specific mutations in a given sequence data. In this context, it is also required to check the sequence which does not correspond to any branch in the phylogeny (haplogroup) or shows discordance between expected and estimated haplogroups, because this can suggest the possible errors including contamination or sample mix-up during sequence analyses.